
Mark S. Senn, Ingo Loa, Jon P. Wright, and J. Paul Attfield, Phys. Rev. B 85 (2012), 125119
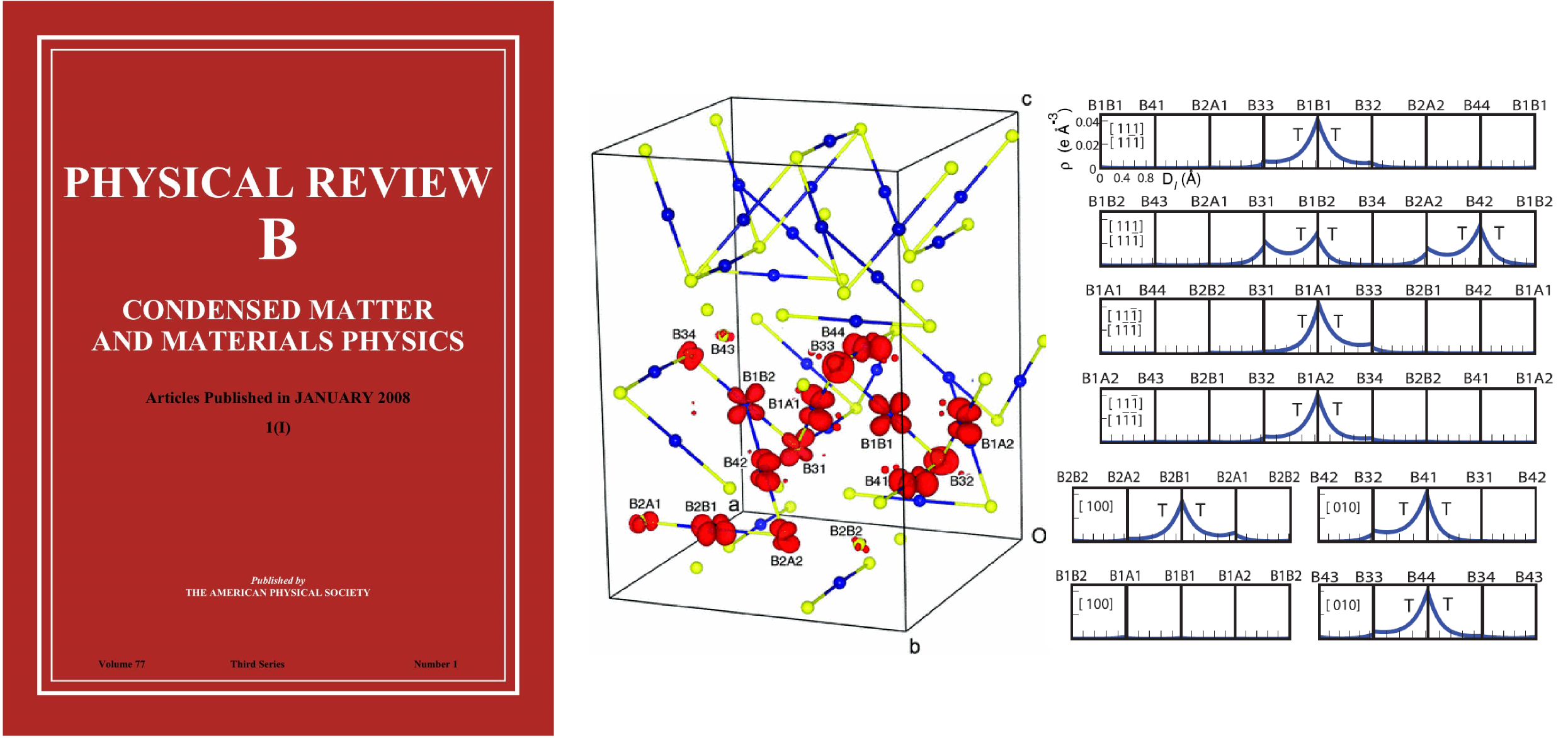
Electronic structure calculations of the Verwey ground state of magnetite, Fe 3 O 4 , using density functional theory with treatment of on-site Coulomb interactions (DFT+U scheme), are reported. These calculations use the recently published experimental crystal structure coordinates for magnetite in the monoclinic space group Cc . The computed density distribution for minority spin electron states close to the Fermi level demonstrates that charge order and Fe 2+ -orbital order are present at the B -type lattice sites to a first approximation. However, Fe 2+ /Fe 3+ charge differences are diminished through weak bonding interactions of the Fe 2+ states to specific pairs of neighboring iron sites that create linear, three-B -atom trimeron units that may be regarded as orbital molecules. Trimerons are ordered evenly along most Fe atom chains in the Verwey structure, but more complex interactions are observed within one chain.
Leave a Comment